Usher Protein Dynamics in Hair Cell Function and Deafness
A major focus of our research is to unravel the mechanisms involved in the trafficking, assembly, and function of stereociliar proteins and to understand how these processes are affected in hearing loss.
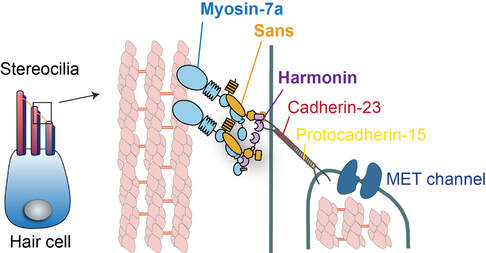
Hair cell stereocilia are a distinctive class of actin-rich membrane protrusions with the ability to convert sound-induced vibrations into electrical signals. At the heart of hair cell mechanotransduction is the tip link, a cadherin-based filament that connects the MET channel to the taller neighboring stereocilium. When the hair bundle deflects, the tip link conveys force to open the MET channel. Actin-based motor myosin-7a is localized at the upper end of the tip link and regulates the tip link's resting tension and hair cell mechanosensitivity. The tail of myosin-7a interacts with two scaffold proteins: sans and harmonin-b. They physically couple the intracellular domain of this link cadherins to the myosin-7a motor and actin core.
Genetic defects of any of the five proteins: myosin-7a (USH1B), sans (USH1G), harmonin-b (USH1C) as well as the tip link proteins cadherin-23 (USH1D) and prtocadherin-15 (USH1F) are causative for Usher syndrome type 1, the most severe form deaf-blindness in humans. Patients with the Type 1 Usher syndrome display profound congenital hearing loss, vestibular dysfunction, and childhood-onset vision loss, underscoring the importance of Usher-1 proteins in the sensory processes.
Our previous research has made major contribution to myosin-7a-based transport, where we detailed the molecular process by which myosin-7a undergoes cargo-dependent dimerization and form a processive transporter capable of moving for long range along actin. Our current research focuses on human myosin-7a holoenzyme and determine its roles in Usher protein trafficking and function during the hearing process.
Hair cell stereocilia are a distinctive class of actin-rich membrane protrusions with the ability to convert sound-induced vibrations into electrical signals. At the heart of hair cell mechanotransduction is the tip link, a cadherin-based filament that connects the MET channel to the taller neighboring stereocilium. When the hair bundle deflects, the tip link conveys force to open the MET channel. Actin-based motor myosin-7a is localized at the upper end of the tip link and regulates the tip link's resting tension and hair cell mechanosensitivity. The tail of myosin-7a interacts with two scaffold proteins: sans and harmonin-b. They physically couple the intracellular domain of this link cadherins to the myosin-7a motor and actin core.
Genetic defects of any of the five proteins: myosin-7a (USH1B), sans (USH1G), harmonin-b (USH1C) as well as the tip link proteins cadherin-23 (USH1D) and prtocadherin-15 (USH1F) are causative for Usher syndrome type 1, the most severe form deaf-blindness in humans. Patients with the Type 1 Usher syndrome display profound congenital hearing loss, vestibular dysfunction, and childhood-onset vision loss, underscoring the importance of Usher-1 proteins in the sensory processes.
Our previous research has made major contribution to myosin-7a-based transport, where we detailed the molecular process by which myosin-7a undergoes cargo-dependent dimerization and form a processive transporter capable of moving for long range along actin. Our current research focuses on human myosin-7a holoenzyme and determine its roles in Usher protein trafficking and function during the hearing process.
Molecular Mechanism of Kinesin-2 in Photoreceptor Trafficking

Cilia are microtubule-based organelles that play crucial roles in the sensory systems including vision. In the neuroretina, the outer segment (OS) of photoreceptors are unique light-sensitive cilia that detect and transduce light into neurological signals. Genetic defects in cilia cause a group of inherited conditions, known as ciliopathies, with clinical manifestations often including retinal dystrophy and blindness.
The assembly and function of cilia requires intraflagellar transport (IFT), a bidirectional movement along cilia powers by motor-IFT complexes. Kinesin-2 motors drive the anterograde IFT and ciliogenesis. Its defects are associated with syndromic and non-syndromic retinal ciliopathies in human patients.
Our research seeks to understand the molecular mechanism by which kinesin-2 transport IFT train along ciliary axoneme, and to decipher how its mutations cause the molecular defects that lead to ciliopathies and retinitis pigmentosa. We address these questions using a combination of biochemical, structural, single-molecule, and modern microscopic approaches.
The assembly and function of cilia requires intraflagellar transport (IFT), a bidirectional movement along cilia powers by motor-IFT complexes. Kinesin-2 motors drive the anterograde IFT and ciliogenesis. Its defects are associated with syndromic and non-syndromic retinal ciliopathies in human patients.
Our research seeks to understand the molecular mechanism by which kinesin-2 transport IFT train along ciliary axoneme, and to decipher how its mutations cause the molecular defects that lead to ciliopathies and retinitis pigmentosa. We address these questions using a combination of biochemical, structural, single-molecule, and modern microscopic approaches.